
High-throughput sequencing has become a cornerstone of biological and medical research. Among its many applications, the vast majority fall under the category of resequencing — that is, aligning sequenced reads to a known reference genome to infer sample composition. Typical resequencing applications include single nucleotide variant (SNV) detection, non-invasive prenatal testing (NIPT), RNA-seq, and metagenomic sequencing. Designing a sequencing method that is both efficient and accurate for such applications is therefore of great importance.
Recently, a team led by Professor Yanyi Huang at Peking University, in collaboration with Cygnus Bio and other partners, published a preprint on bioRxiv titled “A fuzzy sequencer for rapid DNA fragment counting and genotyping.” The study introduces a fuzzy sequencing mode that enables precise alignment of DNA fragments to a reference genome without obtaining the exact four-base DNA sequence, thereby delivering fast and accurate resequencing results.
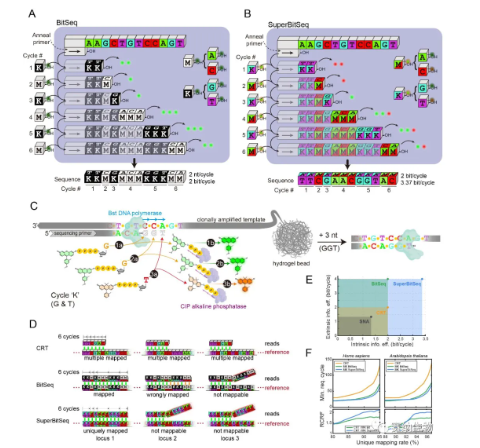
In resequencing data analysis, the most critical step is read alignment to the reference genome. The team first identified that alignment accuracy depends on the information entropy of the sequence and read length. Conventional sequencing produces exact base‑by‑base sequences of four nucleotides, so the information entropy is always twice the read length. A fuzzy sequencing approach could break this linear relationship. To improve alignment accuracy, the goal is to maximize both information entropy and read length within a limited number of sequencing cycles.
Based on this insight, the team proposed two fuzzy sequencing methods. The first, BitSeq, builds on their previously developed fluorogenic sequencing chemistry. In each sequencing cycle, two nucleotides are added together: for example, G and T (collectively called K) in all odd‑numbered cycles, and A and C (collectively called M) in all even‑numbered cycles. The fluorescence signal intensity reveals the number of bases incorporated. After tens of cycles, a binary fuzzy sequence composed of K and M is obtained. Although the resulting sequence is ambiguous, each cycle of BitSeq generates information entropy comparable to that of conventional reversible terminator sequencing (2 bits/cycle), while achieving double the read length. The second method, SuperBitSeq, builds on BitSeq by labeling the two nucleotides in each cycle with different fluorophores. For instance, in odd cycles, G and T are labeled with green and red fluorophores, respectively; in even cycles, A and C are also labeled with green and red. Under this differential labeling, each SuperBitSeq cycle still reads 2 bp/cycle, but the information entropy increases to 3.37 bits/cycle. Simulation‑based alignment experiments using human and Arabidopsis genomes showed that BitSeq and SuperBitSeq require significantly fewer cycles than conventional reversible terminator sequencing to achieve the same unique alignment rate.
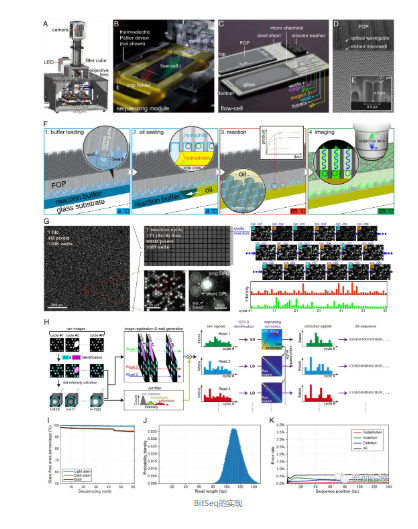
To realize fuzzy sequencing, the team built a complete system encompassing chips, instrumentation, reagents, and algorithms. The chip contains numerous microwells where sequencing reactions take place. The instrument controls reagent flow over the chip, initiates reactions by heating, and captures fluorescence signals with a camera. After image processing and phase correction, fuzzy sequences are output. To prevent cross‑diffusion of fluorescent molecules between microwells, the team used oil sealing to isolate each well. Repeated oil‑sealing of millions of microwells posed a major engineering challenge; after extensive optimization, the team achieved over 90% usable area after dozens of sealing cycles.
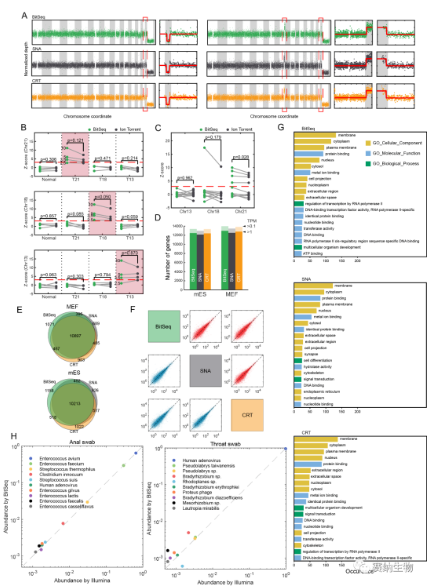
To demonstrate the practicality of BitSeq, the team tested copy number variation (CNV) detection, non‑invasive prenatal testing (NIPT), RNA‑seq, and metagenomic sequencing. The results were highly consistent with those from conventional reversible terminator and semiconductor sequencing methods.
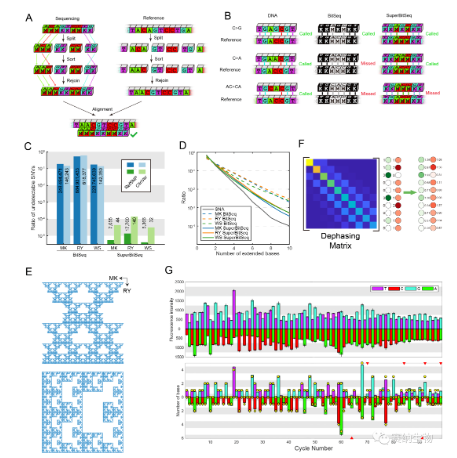
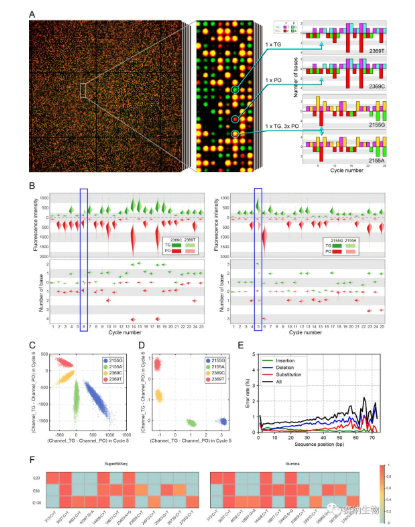
The team also explored the fundamental properties of SuperBitSeq. While BitSeq is well‑suited for counting‑based applications, it cannot detect many types of SNVs. Thanks to its higher information efficiency, SuperBitSeq can detect a much broader range of SNVs. Analysis of the RefSNP and ClinVar databases showed that the proportion of SNVs undetectable by SuperBitSeq is on the order of one in ten thousand or even one in a million, indicating its suitability for the vast majority of SNV detection. Interestingly, the signal output of SuperBitSeq, when encoded, forms a distinctive fractal pattern in a 2D plane, which the researchers named “SuperBitSeq dust.” This fractal structure suggests deeper mathematical principles that merit further investigation.
The team further implemented high‑throughput SuperBitSeq and successfully distinguished the G719S and T790M mutant variants of the EGFR gene from their wild‑type counterparts. They also tested SuperBitSeq on SNV detection in SARS‑CoV‑2 and obtained results consistent with conventional methods.
In summary, this work presents a fuzzy sequencer with two modes — BitSeq and SuperBitSeq — that, within the same number of sequencing cycles, delivers higher information entropy and read length than conventional sequencing methods. By aligning fuzzy sequences directly to a reference genome, the system achieves accurate resequencing analysis without determining the exact four‑base sequence. Multiple application cases, including CNV detection, NIPT, RNA‑seq, metagenomic sequencing, and SNV detection, validate the effectiveness and accuracy of the fuzzy sequencing approach.





